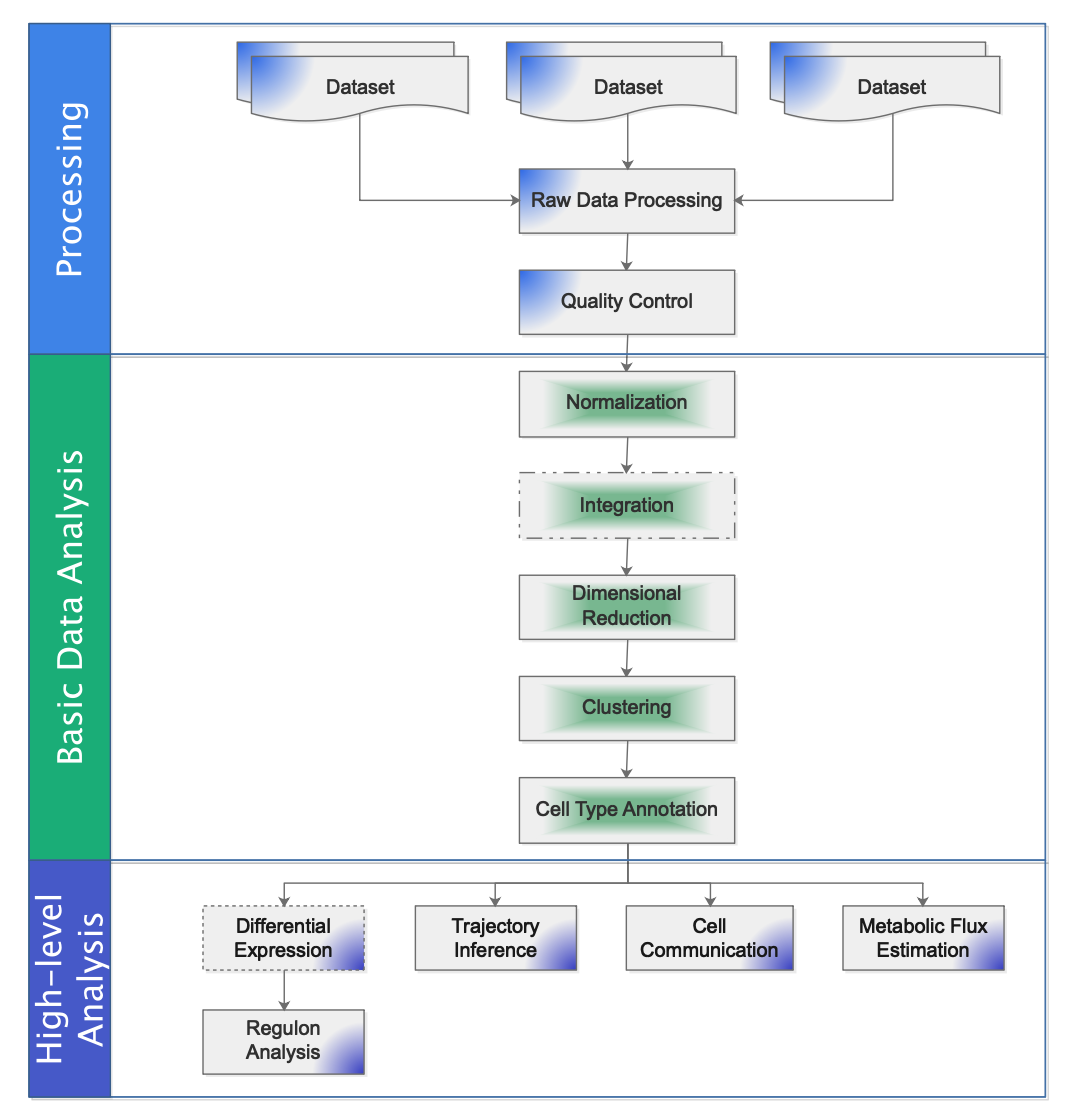
Repo {scrna-recom} 单细胞综述文章关于单细胞转录组分析的流程、软件、安装等的一些说明。这里我主要是拷贝下其中关于核心工具的整理,防止迷失。
Workflow:
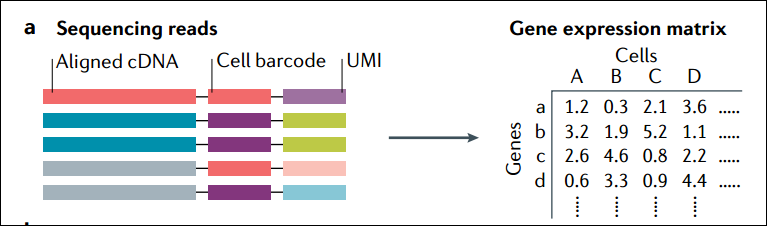
下图展示了从测序read比对参考基因组根据barcode和UMI产生Count matrix的过程。
scRNA-seq利用PCR技术对cDNA分子进行指数级扩增,而UMIs技术可以帮助用户识别并消除在扩增过程中可能产生的重复序列,从而降低技术噪音。值得注意的是,UMI中的测序错误可以人为地提高基因表达,因为应该被消除的重复序列被视为了不同的分子。相反,不同的分子可能会被错误地标记为具有相同的UMI序列,从而被视为同一个分子。
FASTQ 预处理工具:
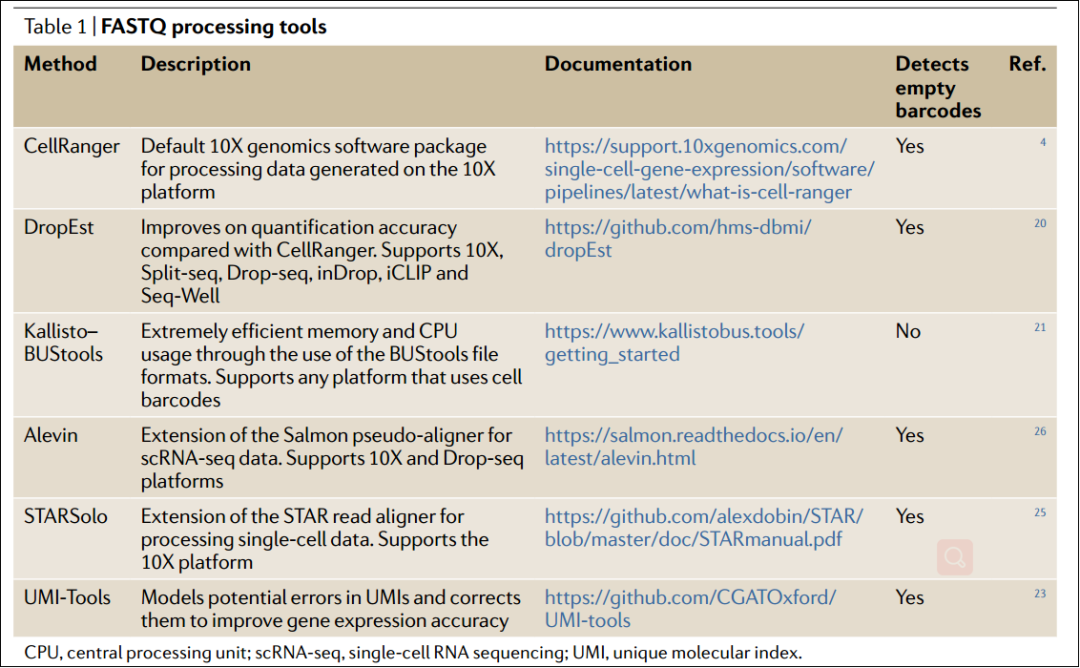
CellRanger、DropEst、Kallisto-BUStools、UMI-Tools、STARSolo和Alevin都是可选的read处理方法,
Cell QC is commonly performed based on three QC covariates:
the number of counts per barcode (count depth):每个细胞的测序深度,gene count数总和
the number of genes per barcode:每个细胞内表达基因的数目
the fraction of counts from mitochondrial genes per barcode:每个细胞中线粒体基因的比例
标准的 R 处理流程大概如下:
Seurat_preprocessing <- function(counts, project = "Scissor_Single_Cell", min.cells = 400, min.features = 0,
normalization.method = "LogNormalize", scale.factor = 10000,
selection.method = "vst", resolution = 0.6,
dims_Neighbors = 1:10, dims_TSNE = 1:10, dims_UMAP = 1:10,
verbose = TRUE){
library(Seurat)
data <- CreateSeuratObject(counts = counts, project = project, min.cells = min.cells, min.features = min.features)
data <- NormalizeData(object = data, normalization.method = normalization.method, scale.factor = scale.factor, verbose = verbose)
data <- FindVariableFeatures(object = data, selection.method = selection.method, verbose = verbose)
data <- ScaleData(object = data, verbose = verbose)
data <- RunPCA(object = data, features = VariableFeatures(data), verbose = verbose)
data <- FindNeighbors(object = data, dims = dims_Neighbors, verbose = verbose)
data <- FindClusters( object = data, resolution = resolution, verbose = verbose)
data <- RunTSNE(object = data, dims = dims_TSNE)
data <- RunUMAP(object = data, dims = dims_UMAP, verbose = verbose)
return(data)
}来源:https://github.com/sunduanchen/Scissor/blob/master/R/Seurat_preprocessing.R
在过去的几年里,UMAP已经取代t-SNE成为scRNA-seq数据的默认可视化方法。与图聚类类似,UMAP生成细胞的最近邻图,根据相似度的强弱对每个细胞间的连接进行加权,然后将图形嵌入到二维空间中。还可以使用PAGA图初始化UMAP,以生成连续开发数据集的高度精确可视化。在实践中,人们发现UMAP在可视化数据集的局部结构方面表现得和t-SNE一样好,包括分离密切相关的细胞类型,同时在可视化数据的全局属性方面表现得更好。因此,对于大多数用户来说,UMAP是一个非常有用的默认可视化选项。对UMAP和t-SNE的其他测试表明,初始化这些方法的方式对它们的整体性能非常重要。实际上,在使用PCA初始化时,t-SNE和UMAP在保存全局结构方面表现得同样好。